Por Bonell Patiño-Escobar, MDInternista-Hematólogo. Post-doctoral fellow, University of California, San Francisco (UCSF). Miembro de AIpocrates.
Durante los últimos años, la conversación sobre inteligencia artificial en medicina ha estado dominada por temas ya familiares: apoyo diagnóstico, lectura de imágenes, modelos predictivos, automatización de tareas clínicas o procesamiento del lenguaje. Y aunque todo eso ha sido, sin duda, transformador, existe una revolución paralela, menos visible para muchos médicos, pero potencialmente igual o incluso más profunda: el uso de inteligencia artificial para diseñar proteínas completamente nuevas, desde cero, con funciones biológicas definidas (1–4). No estamos hablando únicamente de predecir cómo se pliega una proteína. Estamos hablando de algo mucho más ambicioso: imaginar y construir moléculas que no existen en la naturaleza, pero que podrían convertirse en herramientas terapéuticas del futuro (2–6).
Ese punto merece una pausa. Porque si algo sostiene la arquitectura funcional de la vida son precisamente las proteínas. Son receptores, enzimas, anticuerpos, ligandos, sensores y motores de señalización. Gran parte de la medicina moderna y en particular de la oncología de precisión y la inmunoterapia, descansa sobre ellas. Por eso, la posibilidad de diseñarlas racionalmente representa mucho más que un avance técnico: representa una expansión del repertorio mismo con el que la medicina puede intervenir la biología (3–6).Durante décadas, el diseño de proteínas fue un territorio reservado a enfoques altamente especializados, apoyados en física molecular, simulaciones energéticas y ciclos experimentales lentos. Era un campo fascinante, pero exigente y con frecuencia limitado por su complejidad.
Hoy, ese panorama ha empezado a cambiar de forma drástica gracias a una nueva generación de herramientas impulsadas por inteligencia artificial, entre ellas RoseTTAFold, RFdiffusion, ProteinMPNN, AlphaFold 3 y plataformas automatizadas como BindCraft (2–6).Cada una ha contribuido, a su manera, a mover el campo desde la predicción hacia la creación. Por un lado, herramientas como RoseTTAFold y, posteriormente, RoseTTAFold All-Atom, ampliaron la capacidad de modelar no solo proteínas aisladas, sino ensamblajes biomoleculares más complejos. En paralelo, AlphaFold 3 extendió la predicción estructural a complejos que incluyen proteínas, ácidos nucleicos, pequeñas moléculas, iones y residuos modificados, acercando estas plataformas a escenarios mucho más cercanos a la biología real (2,4,5).
Este avance fue reconocido con el Premio Nobel de Química 2024, otorgado a David Baker por el diseño computacional de proteínas, y a Demis Hassabis y John Jumper por la predicción estructural con AlphaFold, un reconocimiento histórico que, por primera vez, consagró a la inteligencia artificial como herramienta central de un descubrimiento científico de primer orden.».RFdiffusion y la lógica de los modelos de difusión: de imágenes a proteínasPara entender qué hace RFdiffusion y por qué representa un salto conceptual tan significativo, vale la pena detenerse brevemente en la idea que lo sostiene.
Los modelos de diffusion, la misma clase de algoritmos que subyace a herramientas de generación de imágenes como DALL·E o Stable Diffusion, funcionan en dos fases. Primero, un proceso de «ruido progresivo»: se toma una imagen real y, paso a paso, se le añade ruido gaussiano hasta convertirla en estática pura. Luego, una red neuronal aprende a revertir ese proceso: dado un punto de ruido, aprende a eliminarlo iterativamente hasta recuperar una imagen coherente y realista. Una vez entrenada, esa red puede generar imágenes completamente nuevas partiendo directamente del ruido, sin necesitar un original. RFdiffusion aplica esta misma lógica al espacio de las proteínas, pero en lugar de píxeles, opera sobre marcos de referencia en el espacio tridimensional que representan la posición y orientación de cada residuo de aminoácido (Figura 1).
El modelo fue entrenado corrompiendo progresivamente estructuras proteicas reales y aprendiendo a reconstruirlas. Al invertir ese proceso sobre ruido puro y condicionarlo a restricciones específicas, como la presencia de un blanco molecular o una arquitectura deseada, es posible generar estructuras proteicas completamente nuevas y funcionales. El resultado es un modelo generativo que no busca en una base de datos ni modifica secuencias existentes: diseña desde cero, como un arquitecto que parte de una hoja en blanco (Figura 2) (3).
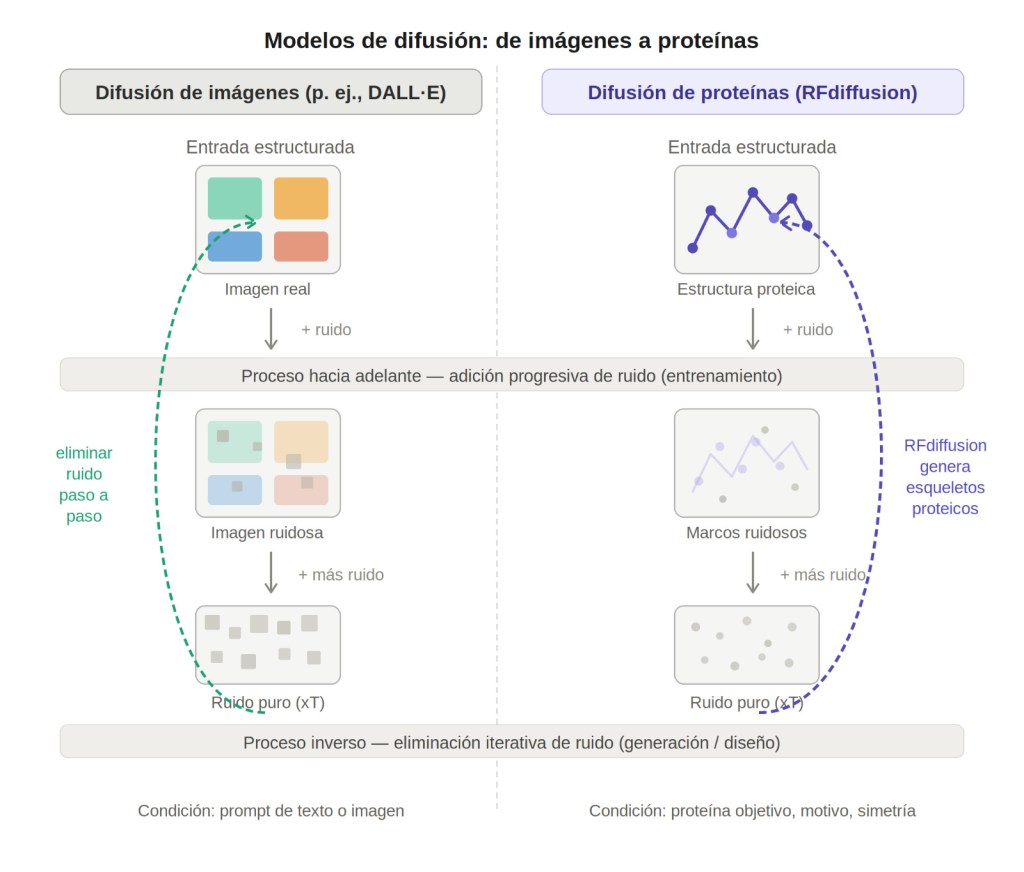
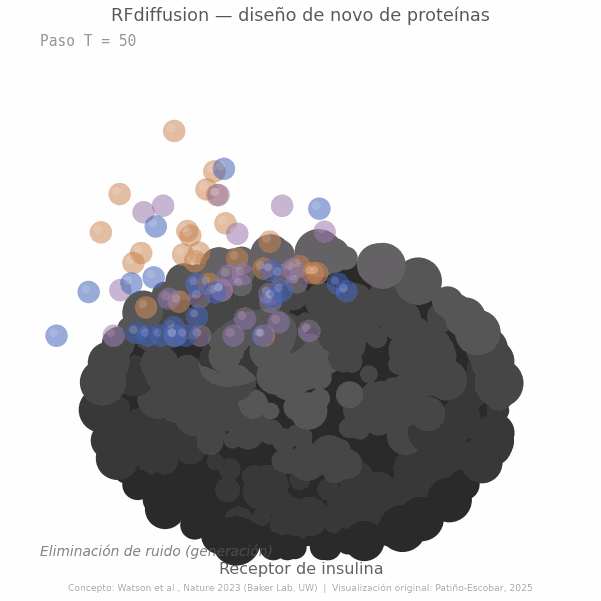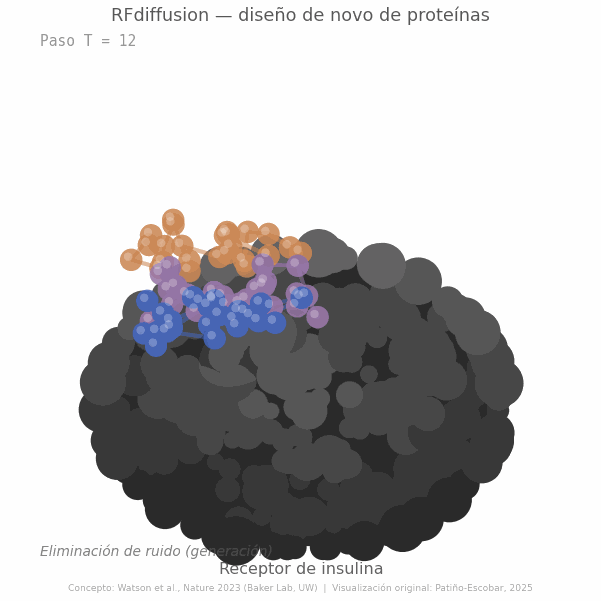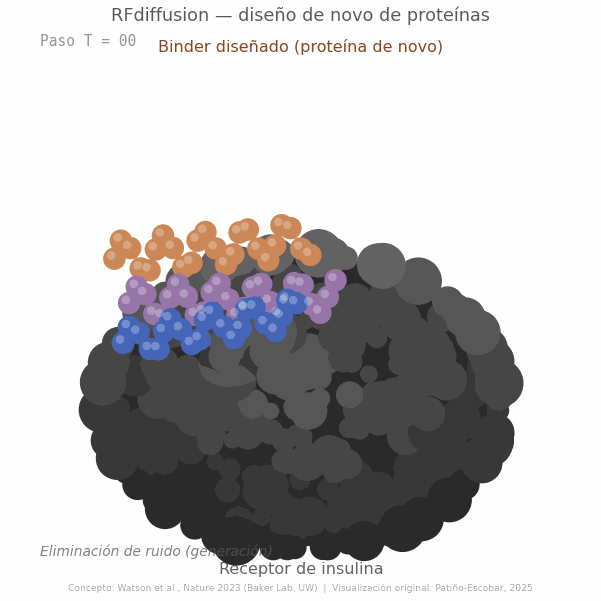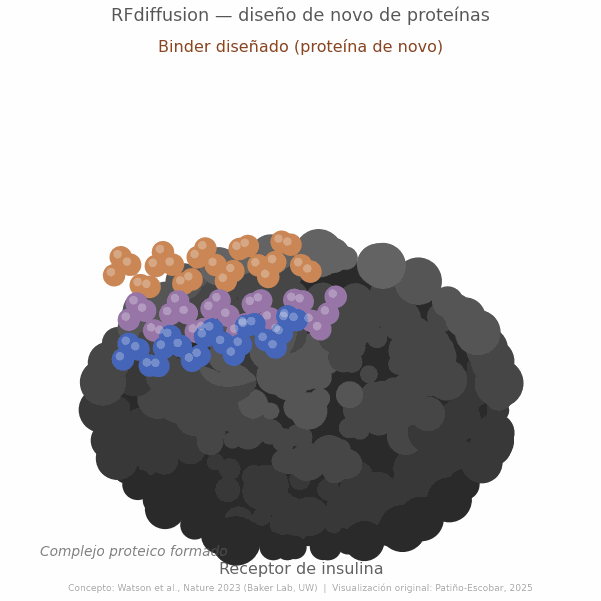
Más tarde, métodos como RFdiffusion llevaron esa lógica un paso más allá: no solo entender la forma, sino generarla, proponiendo arquitecturas proteicas nuevas, scaffolds, interfaces e incluso binders diseñados para reconocer blancos específicos (3). ProteinMPNN, a su vez, ayudó a resolver otro desafío crucial: asignar secuencias de aminoácidos plausibles y estables a esas estructuras diseñadas, acelerando de forma notable el ciclo entre generación, evaluación y validación experimental (4). Y flujos como BindCraft han comenzado a integrar varias de estas capacidades en pipelines automatizados para diseño de binders y mini-binders de novo, con tasas experimentales de éxito reportadas del 10 al 100% según el blanco estudiado (6).
Lo extraordinario de este momento no es solo la sofisticación algorítmica. Es el cambio conceptual que introduce. Durante mucho tiempo, el descubrimiento terapéutico ha dependido, en gran medida, de buscar en la naturaleza, seleccionar entre bibliotecas existentes o modificar moléculas conocidas. Hoy empezamos a entrar en una etapa distinta: una en la que no solo exploramos lo que la biología ya ofrece, sino que comenzamos a proponer nuevas soluciones moleculares con intención y diseño (3–6).
Y pocas áreas podrían beneficiarse tanto de este cambio como la inmunoterapia y la terapia contra el cáncer. La oncología contemporánea es, en buena medida, una ciencia de interfaces: la interfaz entre una célula inmune y una célula tumoral, entre un receptor y su ligando, entre una proteína de superficie y una estrategia terapéutica. Buena parte de nuestras herramientas más exitosas como los anticuerpos monoclonales, anticuerpos biespecíficos, CAR-T, CAR-NK y conjugados fármaco-anticuerpo, dependen, de una u otra forma, de reconocer con precisión una diana biológica y traducir ese reconocimiento en una acción terapéutica.
El diseño computacional de proteínas amplía de manera sustancial ese repertorio, especialmente mediante el desarrollo de binders compactos, modulares y de alta afinidad contra blancos de superficie o interfaces proteína-proteína difíciles de abordar con estrategias convencionales (3,6,7).
En ese contexto, el diseño de proteínas de novo abre posibilidades inmensas. Permite imaginar binders más pequeños, más estables y más programables que los anticuerpos convencionales; módulos de reconocimiento adaptados a epítopos complejos o poco accesibles; ligandos sintéticos capaces de redirigir respuestas inmunes; componentes proteicos para circuitos celulares más refinados; e incluso nuevas estrategias para mejorar direccionamiento, penetración tumoral o especificidad terapéutica (3,6,7).
Para quienes trabajamos cerca de la inmunoterapia, esto resulta particularmente estimulante porque sugiere un futuro en el que no estaremos limitados a reutilizar piezas moleculares ya conocidas, sino que podremos empezar a diseñar las piezas que necesitamos.
Pensemos, por ejemplo, en el desafío cotidiano de encontrar buenos blancos terapéuticos en cáncer. Identificar una diana no siempre se traduce en una herramienta útil para atacarla. Muchas proteínas tumorales presentan superficies difíciles, interacciones débiles o contextos estructurales que complican el desarrollo de anticuerpos o ligandos funcionales. El diseño computacional podría cambiar esa ecuación: partir de la estructura del blanco, generar candidatos de unión, filtrarlos in silico, optimizarlos, validarlos experimentalmente y, a partir de ahí, construir nuevas plataformas terapéuticas. No elimina la biología experimental, por supuesto. Pero sí tiene el potencial de volverla más dirigida, más racional y, en ciertos casos, más rápida (3–6).
Naturalmente, conviene mantener una mirada sobria. El entusiasmo debe ir acompañado de criterio. Diseñar una proteína prometedora en el computador no equivale a tener una terapia lista para el paciente. Entre una estructura elegante y una molécula clínicamente útil existe un trayecto largo, atravesado por problemas de estabilidad, expresión, inmunogenicidad, manufactura, biodistribución, afinidad real en sistemas vivos y seguridad biológica. La validación experimental sigue siendo irremplazable. La biología, afortunadamente, continúa siendo más compleja que cualquier modelo (3,6).
Pero reconocer esas limitaciones no disminuye la magnitud del cambio. Al contrario: la hace más interesante. Porque incluso si hoy estamos apenas viendo los primeros frutos de esta convergencia, ya es evidente que el paradigma se ha desplazado. La inteligencia artificial ha dejado de ser solo una herramienta para clasificar datos clínicos o interpretar imágenes. Está empezando a convertirse en una herramienta para inventar materia biológica funcional (3–6).
Y eso tiene una fuerza transformadora particular en medicina. Porque significa que la IA no solo cambiará la manera en que diagnosticamos, priorizamos o decidimos. También puede cambiar la naturaleza misma de las moléculas con las que tratamos enfermedades.
Desde mi perspectiva, esta es una de las fronteras más fascinantes de la medicina traslacional contemporánea. No solo por su elegancia científica, sino por su potencial real. Imaginar proteínas diseñadas para reconocer tumores con mayor precisión, potenciar células inmunes, bloquear interacciones críticas del microambiente tumoral o servir como nuevas plataformas para terapias celulares ya no pertenece exclusivamente al terreno de la especulación. Poco a poco, empieza a formar parte de una agenda concreta de investigación biomédica (6,7).
Tal vez, en unos años, cuando hablemos del impacto de la inteligencia artificial en oncología, ya no pensemos primero en algoritmos que leen tomografías o predicen pronósticos. Tal vez pensemos en proteínas terapéuticas diseñadas con ayuda de IA, en nuevos binders creados para blancos antes inaccesibles, en inmunoterapias construidas con componentes que no surgieron de la naturaleza, sino de una colaboración inédita entre biología, computación e imaginación humana (3,6,7).
Y quizá ahí resida lo más fascinante de todo: que la inteligencia artificial no solo está transformando cómo entendemos la medicina, sino también qué tipo de medicina somos capaces de crear (1,3–6).
Referencias
- Patiño-Escobar B. La inteligencia artificial y el cambio en el paradigma de la práctica clínica. AIpocrates. 2024.
- Baek M, DiMaio F, Anishchenko I, et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373(6557):871–876.
- Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, et al. De novo design of protein structure and function with RFdiffusion. Nature. 2023;620(7976):1089–1100.
- Dauparas J, Anishchenko I, Bennett N, et al. Robust deep learning–based protein sequence design using ProteinMPNN. Science. 2022;378(6615):49–56.
- Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature. 2024;630:493–500.
- Pacesa M, Nickel L, Schellhaas C, Schmidt J, et al. One-shot design of functional protein binders with BindCraft. Nature. 2025;646:483–492.
- Yang W, Hicks DR, Ghosh A, et al. Design of high-affinity binders to immune modulating receptors for cancer immunotherapy. Nat Commun. 2025;16:2001.
Nota: Esta columna fue escrita «a dos manos», término acuñado por el Dr. Luis Pino, para referirse a la colaboración entre el autor y la inteligencia artificial como herramienta de escritura y asistencia intelectual.
