Autor: Ivanna Frías. Miembro de Aipocrates
Hace algunos meses reflexionábamos en este mismo espacio, en la columna “Más allá del código: la genómica en la era de los datos masivos”, sobre cómo la genómica moderna se ha convertido en una disciplina profundamente marcada por el análisis de datos. Las tecnologías de secuenciación de nueva generación (NGS) han permitido generar cantidades sin precedentes de información genética, revelando millones de variantes a lo largo del genoma humano. Sin embargo, como ocurre con frecuencia en la biología contemporánea, obtener los datos fue solo el primer paso; el verdadero desafío sigue siendo interpretarlos.
En la práctica de la genética clínica, este reto se hace evidente cada vez que solicitamos un estudio genómico. Ya sea mediante paneles multigénicos, secuenciación del exoma o incluso del genoma completo, los resultados suelen ubicarse en uno de tres escenarios posibles. El primero es el más esperado: la identificación de una variante patogénica o probablemente patogénica que permita explicar el cuadro clínico del paciente y establecer un diagnóstico molecular. El segundo escenario es un resultado negativo, en el que no se identifican variantes con relevancia clínica clara.
Las Variantes Genéticas
Para comprender mejor este problema es necesario considerar cómo se clasifican actualmente las variantes genéticas. Durante muchos años fue común utilizar términos como “mutación” o “polimorfismo” para describir cambios en la secuencia del ADN. Sin embargo, estos conceptos podían inducir interpretaciones erróneas, ya que “mutación” solía asociarse automáticamente con enfermedad y “polimorfismo” con variación benigna. Por esta razón, las guías publicadas en el 2015 por el American College of Medical Genetics and Genomics (ACMG) y la Association for Molecular Pathology (AMP) recomendaron adoptar el término “variante” para referirse a cualquier cambio en la secuencia del ADN.
Estas guías establecieron además un sistema estandarizado de clasificación de variantes basado en la evidencia disponible, que las agrupa en cinco categorías:
- Variantes patogénicas.
- Variantes probablemente patogénicas.
- Variantes de significado incierto.
- Variantes probablemente benignas.
- Variantes benignas.
Esta clasificación integra múltiples líneas de evidencia, incluyendo datos poblacionales, estudios funcionales, análisis de segregación familiar y predicciones computacionales.
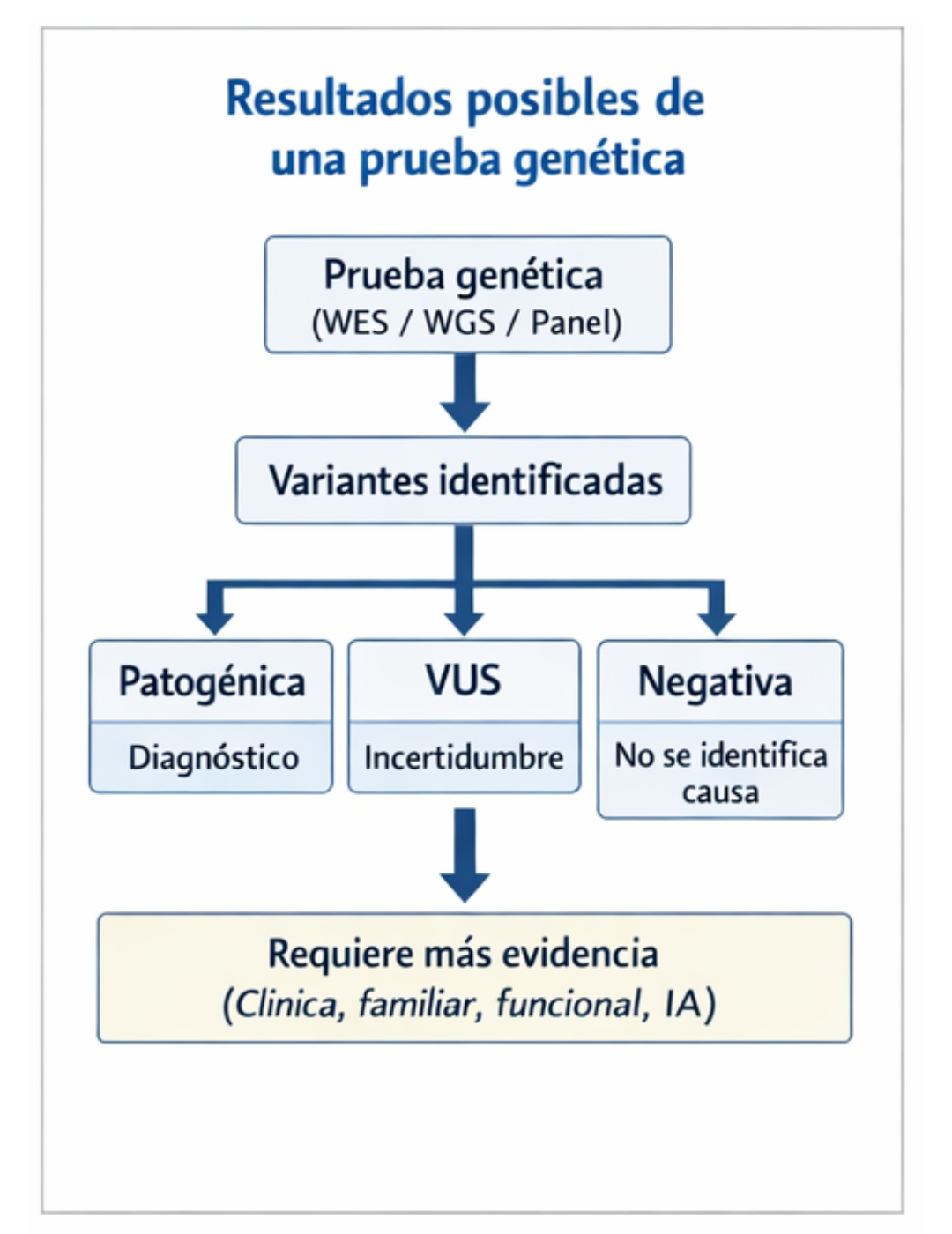
A pesar de este marco estandarizado, una proporción considerable de variantes identificadas mediante secuenciación masiva continúan clasificándose como VUS. Para el genetista clínico, y para otros especialistas involucrados en la interpretación y asesoramiento genético, estas variantes representan uno de los mayores retos de la medicina genómica. Sabemos que el cambio en el ADN existe, pero no podemos afirmar con certeza si es responsable del fenotipo del paciente.
En muchos casos, la pregunta fundamental es aparentemente simple: ¿qué ocurre con la proteína cuando una variante altera la secuencia de un gen?
Responder esta pregunta ha sido históricamente uno de los mayores desafíos de la biología molecular. Las proteínas adoptan estructuras tridimensionales complejas que determinan su función dentro de la célula (Anfinsen, 1973), y pequeñas modificaciones en la secuencia de aminoácidos pueden alterar su estabilidad, su plegamiento o su interacción con otras moléculas (Berg et al., 2015). Durante décadas, estudiar estas estructuras dependía de métodos experimentales complejos como la cristalografía de rayos X o la microscopía crioelectrónica, técnicas altamente especializadas que requieren tiempo y recursos considerables (Callaway, 2020; Jumper et al., 2021).
En los últimos años, sin embargo, el desarrollo de herramientas basadas en inteligencia artificial (IA) ha comenzado a transformar este panorama. Modelos computacionales avanzados tienen la capacidad para predecir la estructura tridimensional de proteínas a partir de su secuencia, permitiendo explorar de manera más rápida cómo ciertas variantes podrían afectar su función (Callaway, 2020; Jumper et al., 2021)
Aunque estas herramientas no sustituyen los estudios funcionales experimentales, representan una nueva capa de evidencia que puede ayudar a comprender el posible impacto de variantes genéticas. En el contexto de la genética clínica, esta información estructural puede aportar pistas valiosas sobre si un cambio en la secuencia podría desestabilizar una proteína, alterar un dominio funcional o modificar su interacción con otras moléculas.
De esta manera, la IA puede abordar uno de los problemas centrales de la medicina genómica: interpretar el significado biológico de las variantes genéticas, particularmente las VUS.
El dilema de las VUS
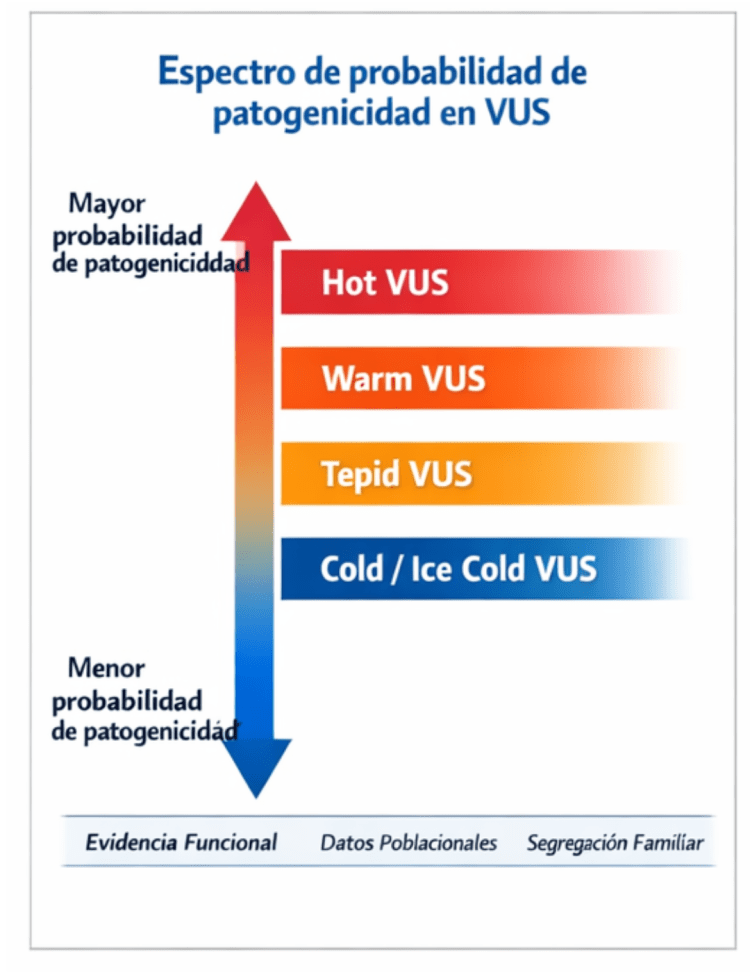
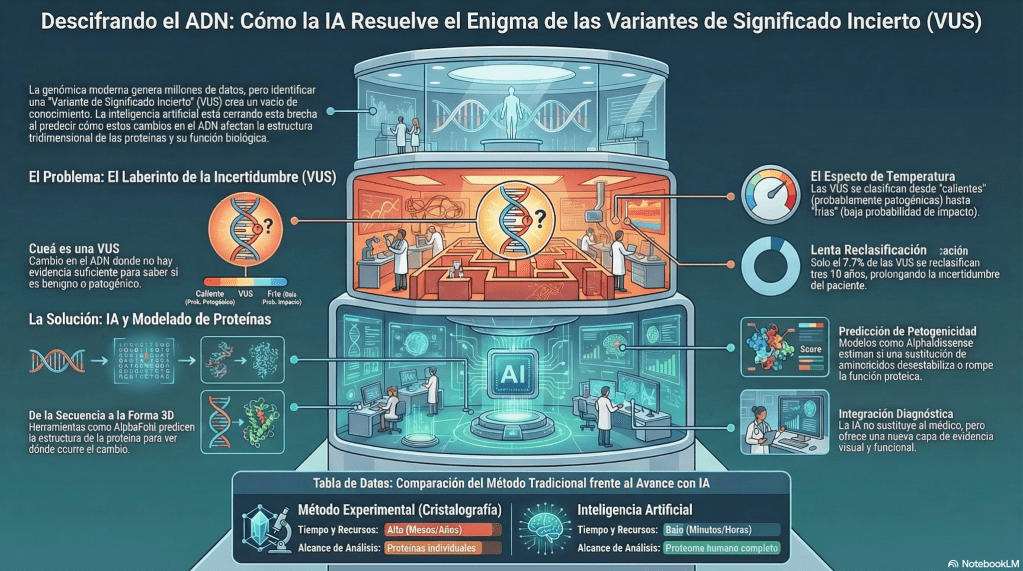
Las VUS no representan una categoría uniforme. En realidad, pueden ubicarse a lo largo de un espectro de probabilidad respecto a su posible impacto patogénico. De forma simplificada, se han descrito distintas categorías dentro de este continuo (Joynt et al., 2021) (Imagen 2):
- VUS “calientes” (hot VUS): cuentan con varias líneas de evidencia que sugieren un posible efecto biológico relevante y podrían reclasificarse como patogénicas si se obtiene información adicional.
- VUS “templadas” (warm VUS): presentan indicios que apuntan hacia una posible relación con el fenotipo clínico, aunque en muchos casos resulta difícil reunir evidencia suficiente que confirme su relevancia.
- VUS “tibias” (tepid VUS): muestran datos limitados o incluso contradictorios respecto a su posible efecto funcional. En estos casos, el contexto clínico del paciente y estudios adicionales, como el análisis de segregación familiar, pueden aportar información útil para clarificar su significado.
- VUS “frías” o “muy frías” (cold/ice cold): constituyen la mayoría de las variantes clasificadas como VUS y presentan una baja probabilidad de reclasificación hacia patogenicidad a medida que se acumula nueva evidencia.
Aunque se espera que la mayoría de las VUS acaben siendo reclasificadas como benignas o probablemente benignas, este proceso suele ocurrir con demasiada lentitud como para tener un impacto inmediato en la atención del paciente (Burke et al., 2022). En un estudio que analizó resultados de pruebas genéticas relacionadas con cáncer durante un periodo de diez años en un gran laboratorio clínico, únicamente el 7.7 % de las VUS únicas fueron reclasificadas en ese intervalo (Mersch et al., 2018).
Mientras tanto, tanto los clínicos como los pacientes suelen encontrar difícil ignorar estos resultados. Un hallazgo de VUS no resuelve la pregunta clínica que motivó el estudio genético y deja al paciente en una situación de incertidumbre. Aunque la evidencia sugiere que, en general, estos resultados no conducen sistemáticamente a intervenciones médicas innecesarias, se han documentado casos en los que se realizaron procedimientos quirúrgicos, estrategias de vigilancia o pruebas familiares que posteriormente resultaron injustificadas (Murray et al., 2011).
Además de sus implicaciones clínicas, las VUS pueden tener un impacto emocional significativo. Algunos pacientes refieren ansiedad, frustración o inquietud prolongada tras recibir este tipo de resultados (Mighton et al., 2021), e incluso describen sentirse paralizados ante la imposibilidad de saber si la variante identificada tiene relevancia para su salud.
Aun así, muchos pacientes prefieren recibir toda la información generada por las pruebas genómicas, incluso cuando esta es incierta, pues consideran que puede contribuir al conocimiento científico y eventualmente beneficiar a sus familias u otros pacientes.
IA y modelaje de proteínas
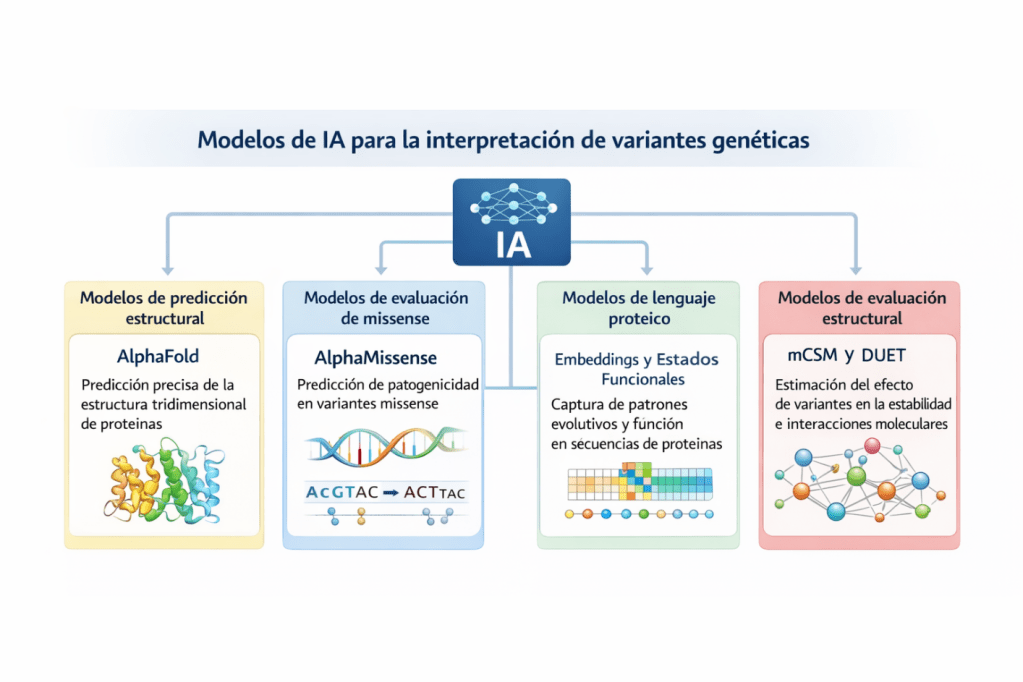
Las herramientas computacionales basadas en IA han comenzado a desempeñar un papel cada vez más relevante en la interpretación de variantes. Algunos de los primeros métodos estructurales desarrollados para predecir el impacto de variantes incluyen SDM (Site Directed Mutator), que utiliza información derivada de familias de proteínas estructuralmente relacionadas para estimar cambios en la estabilidad proteica, y mCSM, que emplea representaciones basadas en grafos para analizar el entorno estructural de un residuo dentro de una proteína. Posteriormente, herramientas integradoras como DUET combinaron diferentes enfoques estructurales y estadísticos para mejorar la precisión en la predicción del efecto de variantes sobre la estabilidad proteica (Pires et al., 2014).
Más recientemente, el desarrollo de modelos de aprendizaje profundo ha ampliado significativamente estas capacidades. Los llamados modelos de lenguaje proteico, basados en arquitecturas tipo transformer, han sido entrenados con millones de secuencias de proteínas y son capaces de capturar patrones evolutivos y funcionales presentes en estas secuencias. Estos modelos generan representaciones matemáticas conocidas como embeddings que pueden utilizarse para predecir el posible impacto de variantes en la función proteica, incluso cuando la información experimental disponible es limitada (Li et al., 2024).
Otro avance fundamental ha sido el desarrollo de modelos de predicción estructural basados en IA. Uno de los más influyentes es AlphaFold, que permite inferir con gran precisión la estructura tridimensional de proteínas a partir de su secuencia de aminoácidos. La disponibilidad de estos modelos estructurales ha ampliado considerablemente la cobertura estructural del proteoma humano, permitiendo analizar cómo determinadas variantes podrían afectar regiones críticas de las proteínas, como dominios funcionales, sitios catalíticos o interfaces de interacción molecular (Jumper et al., 2021).
A partir de estos avances surgieron modelos específicamente diseñados para evaluar el impacto de variantes genéticas. Un ejemplo destacado es AlphaMissense, desarrollado por Google DeepMind, que utiliza información estructural derivada de AlphaFold2 junto con datos de variación genética poblacional para estimar la probabilidad de que una variante missense (cambio en un nucleótido del ADN provoca la sustitución de un aminoácido por otro en la proteína) sea patogénica. Este modelo ha permitido generar predicciones para millones de posibles sustituciones de aminoácidos en proteínas humanas, proporcionando una nueva fuente de evidencia para priorizar variantes candidatas en estudios genómicos (Cheng et al., 2023).
Para facilitar el uso de estas predicciones en análisis genómicos, se han desarrollado herramientas computacionales como AlphaMissenseR, un paquete dentro del ecosistema Bioconductor que permite integrar las predicciones generadas por AlphaMissense con anotaciones genómicas, datos funcionales y bases de datos clínicas como ClinVar. Estas plataformas también permiten visualizar el contexto estructural de las variantes proyectando las predicciones de patogenicidad sobre estructuras tridimensionales de proteínas, lo que facilita la identificación de regiones donde se concentran variantes potencialmente relevantes (Nguyen et al., 2025).
De forma complementaria, se han desarrollado otros enfoques basados en IA para predecir cómo una variante puede afectar la estabilidad estructural de las proteínas o la afinidad de interacción entre biomoléculas. Estos modelos utilizan representaciones tridimensionales de proteínas o grafos moleculares que describen las relaciones espaciales entre los residuos de aminoácidos, lo que permite estimar si una variante podría desestabilizar la proteína o alterar sus interacciones con otras moléculas. De esta manera, ofrecen información valiosa para comprender posibles mecanismos moleculares de enfermedad (Wang et al., 2025).
En conjunto, la integración de herramientas como SDM, mCSM, DUET, AlphaFold, AlphaMissense, AlphaMissenseR y modelos de lenguaje proteico basados en transformers está transformando la forma en que se interpretan las variantes genéticas. Aunque estas aproximaciones computacionales no sustituyen la evidencia clínica ni los estudios funcionales experimentales, sí proporcionan un marco poderoso para priorizar variantes, generar hipótesis biológicas y reducir la incertidumbre asociada (Imagen 3).
Conclusión
Las VUS son el reflejo de una transición en la medicina genómica. Hemos aprendido a leer el genoma con una precisión sin precedentes, pero aún estamos aprendiendo a interpretar su significado.
En este contexto, la integración de la IA, el modelaje estructural de proteínas y el análisis genómico a gran escala está comenzando a transformar este panorama. Estas herramientas permiten conectar, con una precisión creciente, los cambios en la secuencia del ADN con sus posibles consecuencias en la estructura y función de las proteínas (Imagen 4).
Es probable que en los próximos años muchas de las variantes que hoy generan incertidumbre puedan interpretarse con mayor claridad. Mientras tanto, cada VUS pone de manifiesto uno de los principales retos de la genómica contemporánea: transformar la información genética en conocimiento funcional que permita distinguir entre la diversidad genética normal y aquellas variantes con impacto en los mecanismos moleculares de la enfermedad.
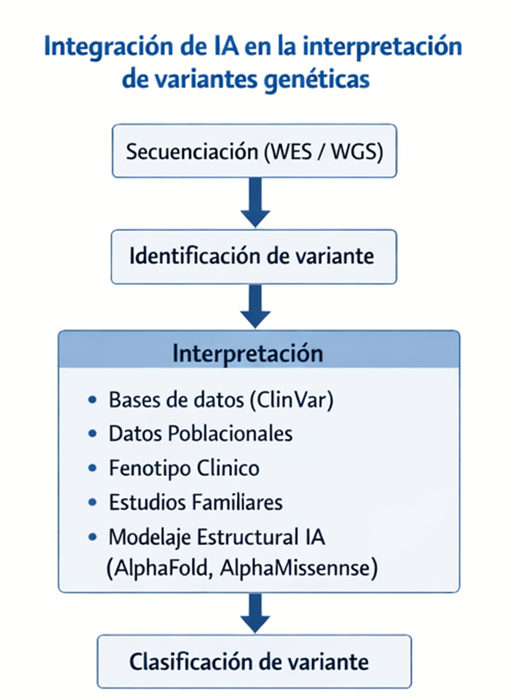
A continuación, una infografía que consolida la información que abordamos en esta columna:
Bibliografía
- Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W. W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K., Rehm, H. L., & ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics, 17(5), 405–424. https://doi.org/10.1038/gim.2015.30
- Anfinsen C. B. (1973). Principles that govern the folding of protein chains. Science (New York, N.Y.), 181(4096), 223–230. https://doi.org/10.1126/science.181.4096.223
- Berg, J. M., Tymoczko, J. L., Stryer, L., & Gatto, G. J. (2019). Biochemistry (9ª ed.). W. H. Freeman and Company.
- Callaway, E. (2020). ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature, 588(7837), 203-204. https://doi.org/10.1038/d41586-020-03348-4
- Jumper, J., Evans, R., Pritzel, A., Kohli, P., & Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583-589. https://doi.org/10.1038/s41586-021-03819-2
- Joynt, A. C. M., Axford, M. M., Chad, L., & Costain, G. (2021). Understanding genetic variants of uncertain significance. Paediatrics & child health, 27(1), 10–11. https://doi.org/10.1093/pch/pxab070
- Burke, W., Parens, E., Chung, W. K., Berger, S. M., & Appelbaum, P. S. (2022). The Challenge of Genetic Variants of Uncertain Clinical Significance: A Narrative Review. Annals of internal medicine, 175(7), 994–1000. https://doi.org/10.7326/M21-4109
- Mersch, J., Brown, N., Pirzadeh-Miller, S., Mundt, E., Cox, H. C., Brown, K., Aston, M., Esterling, L., Manley, S., & Ross, T. (2018). Prevalence of Variant Reclassification Following Hereditary Cancer Genetic Testing. JAMA, 320(12), 1266–1274. https://doi.org/10.1001/jama.2018.13152
- Murray, M. L., Cerrato, F., Bennett, R. L., & Jarvik, G. P. (2011). Follow-up of carriers of BRCA1 and BRCA2 variants of unknown significance: variant reclassification and surgical decisions. Genetics in medicine : official journal of the American College of Medical Genetics, 13(12), 998–1005. https://doi.org/10.1097/GIM.0b013e318226fc15
- Mighton, C., Shickh, S., Uleryk, E., Pechlivanoglou, P., & Bombard, Y. (2021). Clinical and psychological outcomes of receiving a variant of uncertain significance from multigene panel testing or genomic sequencing: a systematic review and meta-analysis. Genetics in medicine : official journal of the American College of Medical Genetics, 23(1), 22–33. https://doi.org/10.1038/s41436-020-00957-2
- Pires, D. E. V., Ascher, D. B., & Blundell, T. L. (2014). mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics, 30(3), 335-342. https://doi.org/10.1093/bioinformatics/btt691
- Pan, Q., Portelli, S., Nguyen, T. B., & Ascher, D. B. (2024). Characterization on the oncogenic effect of the missense mutations of p53 via machine learning. Briefings in Bioinformatics, 25(1), bbad428. https://doi.org/10.1093/bib/bbad428
- Cheng, J., Novati, G., Pan, J., Bycroft, C., Žemgulytė, A., Applebaum, T., Pritzel, A., Wong, L. H., Zielinski, M., Sargeant, T., Schneider, R. G., Senior, A. W., Jumper, J., Hassabis, D., Kohli, P., & Avsec, Ž. (2023). Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science, 381(6664), eadg7492. https://doi.org/10.1126/science.adg7492
- Nguyen, T. N., Lee, T., Turaga, N., Gentleman, R., Geistlinger, L., & Morgan, M. (2025). AlphaMissenseR: An integrated framework for investigating missense mutations in human protein-coding genes. Bioinformatics Advances, 5(1), vbaf093. https://doi.org/10.1093/bioadv/vbaf093
- Lu, Q., Ding, J., Li, L., & Chang, Y. (2025). Graph contrastive learning of subcellular-resolution spatial transcriptomics improves cell type annotation and reveals critical molecular pathways. Briefings in Bioinformatics, 26(1), bbaf020. https://doi.org/10.1093/bib/bbaf020
