Andrés F. Cardona1, Luis Eduardo Pino2, Oscar Arrieta3
1Instituto de Investigación y Educación, Centro de Tratamiento e Investigación sobre Cáncer Luis Carlos Sarmiento Angulo (CTIC), Bogotá, Colombia
2OxLER y AIpocrates, Bogotá, Colombia
3Dirección general, Instituto Nacional de Cancerología – INCaN, Ciudad de México, México
Principales Características Funcionales del Cáncer según Weinberg y Hanahan
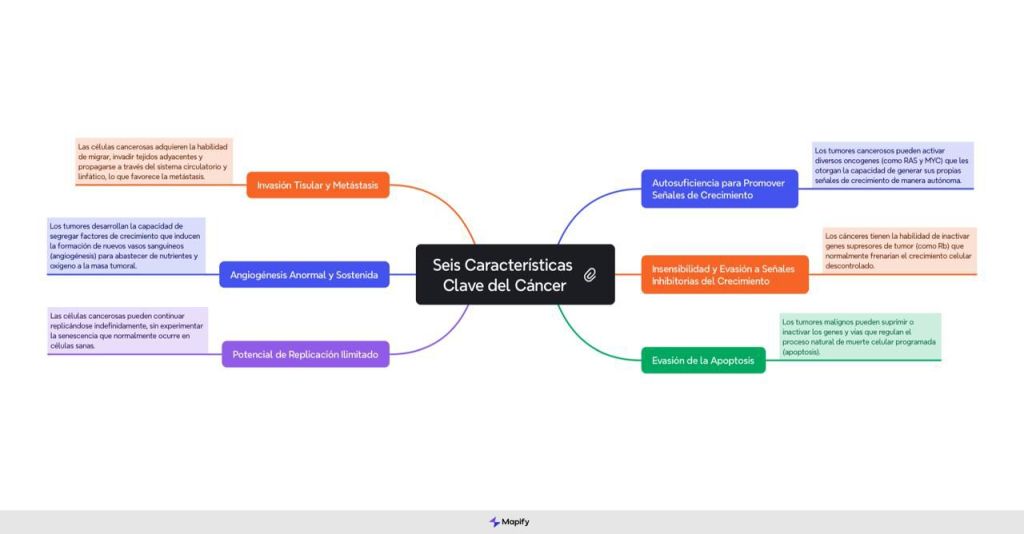
En el año 2000, los biólogos Robert Weinberg y Douglas Hanahan publicaron la primera aproximación editorial a los arquetipos primarios del cáncer (1). En ese entonces, reconocieron que las neoplasias se producen a partir de la acumulación errática de diversas mutaciones y enumeraron seis alteraciones esenciales en la fisiología celular que determinan la evolución de la enfermedad. La autosuficiencia para promover la generación de señales de crecimiento a través de un impulso autónomo en virtud de la activación de diversos oncogenes (por ejemplo, RAS y MYC); b. La insensibilidad y evasión a las señales inhibitorias del crecimiento a través de la inactivación de genes supresores de tumor (Rb); c. La evasión sistemática de la apoptosis a través de la supresión e inactivación de los genes y vías que permiten su curso natural; d. El potencial de replicación ilimitado (inmortalización) incluso después de generaciones; e. La angiogénesis anormal y sostenida que se adquiere a través de la segregación de factores de crecimiento; f. La invasión tisular y metástasis que favorecen la capacidad migratoria y de propagación por contigüidad o a través de vasos sanguíneos y linfáticos (2).
Durante años, la ciencia consideró que el cáncer estaba constituido por poblaciones celulares homogéneas (3), con múltiples características comunes y otras particulares al linaje histológico. Sin embargo, el fenotipo tumoral incluye en la actualidad a diversos componentes formes del microambiente tumoral que hacen parte del estroma, capaz de afectar críticamente la evolución de la enfermedad. Parte de los modelos evolutivos del cáncer se explican a través de la transición cualitativa, determinista e irreversible de las células tumorales pluripotenciales. Hoy reconocemos que estas tienen la capacidad de entrar en quiescencia o senescencia sin contribuir positivamente con el crecimiento tumoral. La heterogeneidad espacial y temporal está definida por las causas iniciales de los eventos oncogénicos, por los sucesos qua afectan los sistemas biológicos incluyendo los cambios genéticos, epigenéticos y lisogénicos, por las vías de señalización alteradas, y por las características fenotípicas de las células tumorales que les permiten competir con éxito en el microambiente tumoral para lograr la expansión (3,4).
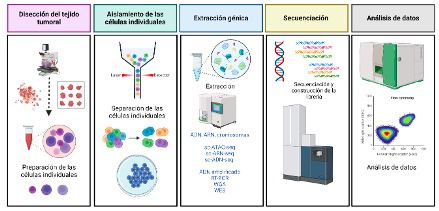
En el campo de la biología celular, el análisis unicelular y subcelular permiten el estudio de la genómica, transcriptómica, proteómica, metabolómica y de las interacciones célula-célula (5). El concepto de análisis unicelular (del inglés, single cell analysis – SCA) se originó en la década de 1970. Antes del descubrimiento de la heterogeneidad tumoral, el análisis unicelular se refería principalmente a la evaluación y manipulación de una célula en una población masiva de ellas utilizando el microscopio óptico y electrónico (6). Actualmente, el SCA de elementos eucariotes y procariotes, permite elucidar diversos mecanismos que promueven la enfermedad, su evolución y resistencia a las intervenciones oncológicas. Tecnologías como la clasificación de células activadas por fluorescencia (FACS) permiten el aislamiento preciso de células individuales seleccionadas de muestras complejas, mientras que las tecnologías de división de elementos formes individuales de alto rendimiento, facilitan el análisis molecular simultáneo de cientos o miles de células individuales sin clasificar. Esto ha permitido la exploración en profundidad del transcriptoma de células genotípicamente idénticas, facilitando la subclasificación celular del cáncer (7). Por otra parte, las técnicas de espectrometría de masas se ha convertido en una herramienta analítica importante para el análisis proteómico y metabolómico de células individuales. Los avances recientes han permitido cuantificar miles de proteínas en cientos de células individuales (8), y, por lo tanto, facilitar la búsqueda de biomarcadores y nuevos medicamentos.
En la actualidad los esfuerzos de la SCA se concentran en el desarrollo de la secuenciación de ADN unicelular (scDNA-seq), la secuenciación de ARN de una sola célula (scRNA-seq), el ensayo de una sola célula para el estado de la cromatina por secuenciación (scATAC-seq), la indexación celular de transcriptomas y epítopos de una sola célula por secuenciación (scCITE-seq), el estudio de accesibilidad a la cromatina de un solo núcleo y la secuenciación de la expresión de ARNm unicelular (scSNARE-seq), etc. (8). La Figura 1 incluye el diagrama de flujo de un estudio típico de secuenciación ómica de una sola célula.
La inteligencia artificial y la biología tumoral
La inteligencia artificial (IA) ha penetrado rápidamente el desarrollo de la biología molecular tumoral. Solo en 2018 se invirtieron 2.8 billones de dólares (USD) en el avance de modelos de aprendizaje profundo (DL; del inglés, deep learning) y aprendizaje automático (ML; del inglés machine learning) para optimizar algoritmos de análisis para scRNA-seq (9). Estos, incluyen pruebas de validación que disponen de la información disponible públicamente en Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/), en el Human Cell Atlas (https://www.humancellatlas.org/), y en 10x Genomics (https://www.10xgenomics.com/resources/datasets) (10).
Recientemente, Sinha y colaboradores publicaron los resultados de la plataforma PERCEPTION para predecir la respuesta y resistencia al tratamiento a través de un análisis avanzado de transcriptómica en células individuales (11). Este estudio como prueba de concepto utilizó la ML por transferencia para entrenar un modelo que permite predecir la repuesta a medicamentos a partir del análisis de la secuenciación masiva de ARN ajustados por scRNA-seq. El modelo incluyó 44 medicamentos aprobados previamente por la FDA (Federal Drug Administration) que se probaron en
Figura 1. Diagrama de flujo para un proyecto de secuenciación ómica de células individuales. (A) disección de tejido y preparación de suspensiones para las células individuales; (B) aislamiento de las células individuales; (C) extracción de información genética importante con diversas tecnologías para el estudio de células individuales (scDNA-seq, scRNA-seq y sc-ATAC-seq). ScDNA-seq: el ADN se extrae de células individuales utilizando técnicas específicas, seguido de WGA (secuenciación del genoma completo) para obtener la información completa de la secuencia de ADN; ScRNA-seq: el ARN se extrae de células individuales utilizando técnicas particulares, seguido de conversión del ARN por ADNc utilizando transcriptasa inversa seguida de amplificación mediante PCR para obtener toda la información del transcriptoma; y sc-ATAC-seq que utiliza la transposasa Tn5, para capturar la información de la región variable de la cromatina, corta el ADN nuclear para obtener fragmentos del mismo, amplifica por PCR, obtiene toda la información del genoma nuclear de la región abierta de la cromatina.; (D), secuenciación de células individuales mediante una plataforma específica; (E), Análisis de datos.
41 pacientes con mieloma múltiple y 33 con cáncer de mama. El estudio permitió determinar que la presencia de un solo clon resistente predice la resistencia por evolución clonal, incluso si el resto de las células respondían. Además, el modelo predijo con éxito el desarrollo de resistencia en 24 pacientes con cáncer de pulmón de células no pequeñas. Previamente, Chen y colaboradores desarrollaron un modelo de DL (scDEAL) para imputación de la expresión génica a partir de datos masivos provenientes de scRNA-seq (12). El modelo scDEAL fue capaz de predecir la sensibilidad a los medicamentos oncológicos a nivel de una sola célula considerando el genotipo y la expresión de mRNA evaluada en cada célula. El scDEAL fue suficiente para armonizar una enorme cantidad de información permitiendo la transferencia de la respuesta farmacológica a la clínica (12). En adición, el modelo scDEAL infirió los genes asociados con la respuesta y la resistencia a partir de la incorporación de la información incluida en las bases de datos Genomics of Drug Sensitivity in Cancer (GDSC) y Cancer Cell Line Encyclopedia (CCLE). El principal obstáculo para desarrollar una herramienta como el scDEAL es la potencia de entrenamiento insuficiente debido al número limitado de datos de referencia de dominio público.
Dhomen y colaboradores intentaron contestar una pregunta simple a través de un modelo de ML, ¿Es posible generar un modelo clasificador que permita diferenciar correctamente las células tumorales de las normales en múltiples tipos de cáncer? El modelo IKARUS consta de dos pasos, primero, el descubrimiento de una firma celular tumoral completa en forma de un conjunto de genes alterados y agrupados por conjuntos de datos de células individuales revisados por expertos. Segundo, el entrenamiento del clasificador a través de una regresión logística que permitió discriminar las células tumorales y normales por sus perfiles de mRNA seguido de la propagación basada en red usando etiquetas celulares únicas y en red considerando la interacción célula-célula construida a la medida (13). Con él objetivo de desarrollar un clasificador in silico robusto, sensible y reproducible se probó el IKARUS en múltiples conjuntos de datos de células individuales obtenidos por diferentes técnicas de secuenciación utilizando un procedimiento de dos pasos. El primero, uso el análisis de expresión diferencial enriquecido con los genes conocidos y relacionados con diversos canceres. Para obtener la lista de genes y realizar su validación cruzada se utilizaron conjuntos de entrenamiento, validación y prueba. De forma simple, el clasificador con el mejor rendimiento combinó el conjunto de datos de cáncer colorrectal con cáncer de pulmón, logrando una precisión mínima de 0.97. La validación se llevó a cabo en el conjunto de genes de carcinoma hepatocelular que obtuvo una precisión e 0.93 (13).
La integración del análisis por scRNA-seq para la búsqueda de nuevos medicamentos dará su siguiente paso a través de las redes generativas antagónicas (GAN); por ejemplo, el desarrollo de la plataforma GENTRL (modelo de aprendizaje de refuerzo tensorial generativo) ha permitido el diseño de nuevos inhibidores de tirosin-quinasa, priorizando las estructuras a partir de la viabilidad sintética, la innovación y su actividad biológica (Figura 2). El modelo GENTRL impulsó el descubrimiento del fármaco INS018_055 que ya se encuentra en pruebas clínicas para el manejo de la fibrosis pulmonar idiopática (IPF) (14). Para desarrollar la hipótesis inicial se utilizó el módulo de descubrimiento de PandaOmics y la información de CHEMISTRY42. El proceso se entrenó con una colección de conjuntos de datos ómicos que incluyó scRNA-seq y clínicos relacionados con la fibrosis pulmonar. La selección de objetivos se realizó utilizando un sofisticado sistema de puntuación para los genes y sus vías derivado del iPANDA (análisis de descomposición de redes de activación de vías in silico) en 2016. PandaOmics identificó puntos relevantes mediante el análisis de síntesis profunda usando inferencia de causalidad y reconstrucción de vías de novo para generar una lista de genes potenciales. Un motor de procesamiento del lenguaje natural (PLN) evaluó la innovación mediada por el blanco molecular potencial y lo asoció con la puntuación para validar su relevancia. Este apartado incluyó datos de millones de archivos incluyendo patentes, publicaciones, subvenciones y bases de ensayos clínicos (14). Fue así como PandaOmics reveló 20 objetivos potenciales, y seleccionó un gen que codifica una proteína intracelular que interactúa con las quinasas TRAF2 y NCK (TNIK). En este caso, la literatura previa lo había vinculado tangencialmente con múltiples vías de señalización promotoras de la FPI, a saber, las rutas wingless/b-catenina (WNT), del factor transformante beta (TGF-β), Hippo, c-Jun N-terminal kinase (JNK) y del factor nuclear kappa B (NF-κB). Sin embargo, TNIK nunca se había estudiado como un objetivo terapéutico en la FPI y, por lo tanto, fue altamente clasificado por el algoritmo de IA. La asociación de TNIK con la FPI se validó utilizando los conjuntos de datos de expresión génica de células individuales de pulmón sano y tejido pulmonar fibrótico de pacientes con FPI, con los que confirmamos su enriquecimiento en el tejido patológico (14). A continuación, se utilizó el módulo de química generativa CHEMISTRY42 que diseñó el medicamento más específico para inhibir TNIK. Para este fin, CHEMISTRY42 utiliza un flujo de trabajo basado en la estructura (SBDD), que se utilizó para generar una librería de estructuras virtuales potenciales.
Figura 2. Desde el inicio del proyecto en 2019 hasta el ensayo clínico fase 2A para pacientes con FPI. Esta cronología cubre el alcance completo del estudio impulsado por IA; tras su priorización de indicaciones en 2020, el INS018_055 fue validado como un inhibidor de TNIK potente, seguro y selectivo después del análisis in vitro. Su eficacia se probó en tres modelos de fibrosis murina in vivo, pasó los estándares de seguridad en estudios fase 0 y fase 1, y se está probando en dos ensayos clínicos fase 2A en EE. UU. y China.
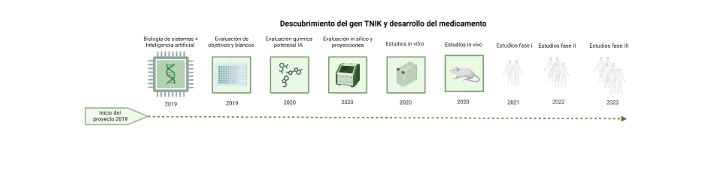
Se emplearon treinta modelos generativos en paralelo para producir las estructuras de los compuestos, tras lo cual un grupo de científicos proporcionaron comentarios sobre la optimización de esta selección virtual. Tras múltiples iteraciones, se seleccionó el sitio de unión de ATP de TNIK como el bolsillo de unión objetivo. Inicialmente, el producto se llamó ISM001 y demostró actividad biológica con un valor de concentración inhibitoria máxima (CI50) nanomolar medio contra TNIK. Este paso de generación de compuestos de novo optimizó aún más el diseño del INS018_055 para aumentar la solubilidad, promover un buen perfil de seguridad y conservar su notable actividad inhibitoria contra TNIK (14,15). Es importante destacar que los investigadores convirtieron una teoría basada en una hipótesis plausible en una realidad convincente que permitió producir modelos animales optimizados (tratamiento oral o inhalado en ratones con FPI) donde se determinó que el ISM001 reduce la activación de los fibroblastos, la deposición de proteínas profibróticas y atenuar la inflamación pulmonar crónica mejorando sí la función pulmonar. Además, el INS018_055 demostró una función inhibidora panfibrótica, atenuando el daño renal, cutáneo y cardíaco en dos modelos in vivo adicionales. Esta observación favoreció las oportunidades de expansión de la indicación para la molécula y será un área de investigación futura. Tras la conclusión de la fase preclínica el INS018_055 logró la nominación como medicamento candidato a estudio a principios de 2021, aproximadamente, 18 meses después de la propuesta de PandaOmics para TNIK como un objetivo farmacológico para la FPI en 2019.
En noviembre de 2021, solo nueve meses después de su nominación, se anunció que los primeros voluntarios sanos habían recibido la dosis base en un ensayo de microdosis en humanos (FIH) de INS018_055 en Australia (ACTRN12621001541897). Este estudio se llevó a cabo en ocho voluntarios sanos, superó las expectativas y demostró perfiles farmacocinéticos y de seguridad favorables para el INS018_055. Estos datos prepararon el terreno para el siguiente paso de las pruebas clínicas. Es importante destacar que este éxito supuso un hito enorme para el campo más amplio de la IA generativa: desde el descubrimiento de nuevos objetivos terapéuticos hasta las pruebas en el estudio fase 1 en menos de 30 meses. En relación con la metodología tradicional para el descubrimiento de fármacos, este enfoque impulsado por IA logró esta hazaña en la mitad del tiempo convencional y por una fracción del costo.
Tras estos resultados positivos, el desarrollador colaboró con investigadores de Nueva Zelanda para llevar a cabo el ensayo clínico fase 1 aleatorizado, doble ciego y controlado con placebo (NCT05154240) con la intención de evaluar la seguridad, la tolerabilidad y las propiedades farmacocinéticas del INS018_055 en 78 voluntarios sanos. Este estudio temprano se inició en febrero de 2022 y la visita de seguimiento final se completó en noviembre del mismo año. Los voluntarios sanos se inscribieron en diez cohortes de 5 subgrupos tratados con una dosis única ascendente (SAD) y 3 subgrupos de dosis múltiple ascendente (MAD) más otros 14 voluntarios sanos para el estudio de interacción fármaco-fármaco (DDI) que permitió determinar la dosis máxima tolerada y establecer pautas de dosificación para el estudio fase 2. En enero de 2023, se concluyó que los datos farmacocinéticos en humanos del INS018_055 coincidían totalmente con el modelo preclínico, sin acumulación indeseable del fármaco después de un período de siete días posterior al inicio del tratamiento. Además, el INS018_055 fue seguro y bien tolerado, sin eventos adversos graves ni mortalidad reportados en ninguno de los participantes sanos. Todos los efectos adversos relacionados con el tratamiento en las cohortes SAD y MAD fueron en el informe final. Un segundo estudio fase 1 en una población diferente en China (CTR20221542) concluyó hallazgos similares. Los dos estudios tempranos coincidieron en que el INS018_055 fue seguro, bien tolerado y posee una buena biodisponibilidad oral y farmacocinética proporcional a la dosis en voluntarios sanos. Luego se iniciaron dos estudios fase 2A usando la dosis seleccionada para el INS018_055 (NCT05975983 y NCT05938920) en pacientes con FPI. En curso del reclutamiento de 120 pacientes en más de 40 sitios de investigación diferentes, se pretende evaluar la seguridad, tolerabilidad y eficacia durante 12 semanas de tratamiento. El éxito del INS018_055 en los estudios preclínicos y ensayos clínicos de fase temprana tiene implicaciones significativas para la investigación. De hecho, estos logros validan la eficacia de una suite (Pharma AI) para el descubrimiento de fármacos con IA y la naturaleza integral de su recorrido. Estos resultados también sientan un precedente para explorar el potencial de esta tecnología para acelerar el descubrimiento de fármacos en muchos otros contextos, a una fracción del costo y el tiempo del método tradicional.
Esta expansión abordaría directamente muchos de los desafíos que enfrenta la investigación y desarrollo de la industria farmacéutica. Por ejemplo, los esfuerzos para el descubrimiento e identificación de objetivos dirigidos por IA en cáncer pueden reducir la probabilidad de que un objetivo identificado no llegue a ser nominado como candidato preclínico. Este costoso paso que emprenden la academia y el sector privado es un gran obstáculo para muchos proyectos de investigación. Evitar objetivos erróneos o esfuerzos redundantes podría ser un área en la que la IA dejaría una huella duradera. Además, los avances computacionales en velocidad y potencia seguirán agilizando drásticamente la generación de nuevas moléculas a través de herramientas como CHEMISTRY42. La aplicación de la IA en el estudio de célula única en cáncer se extiende a la IA generativa como factor primario para revolucionar el descubrimiento y la promoción de nuevas terapias dirigidas.
Referencias
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57-70. doi: 10.1016/s0092-8674(00)81683-9.
- Floor SL, Dumont JE, Maenhaut C, Raspe E. Hallmarks of cancer: of all cancer cells, all the time? Trends Mol Med. 2012 Sep;18(9):509-15. doi: 10.1016/j.molmed.2012.06.005.
- Kumar V, Stewart JH 4th. Immunometabolic reprogramming, another cancer hallmark. Front Immunol. 2023 May 19;14:1125874. doi: 10.3389/fimmu.2023.1125874.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74. doi: 10.1016/j.cell.2011.02.013. PMID: 21376230.
- Chen S, Jiang W, Du Y, Yang M, Pan Y, Li H, Cui M. Single-cell analysis technologies for cancer research: from tumor-specific single cell discovery to cancer therapy. Front Genet. 2023 Oct 12;14:1276959. doi: 10.3389/fgene.2023.1276959.
- Han Y, Wang D, Peng L, Huang T, He X, Wang J, et al. Single-cell sequencing: a promising approach for uncovering the mechanisms of tumor metastasis. J Hematol Oncol. 2022 May 12;15(1):59. doi: 10.1186/s13045-022-01280-w.
- Lei Y, Tang R, Xu J, Wang W, Zhang B, Liu J, et al. Applications of single-cell sequencing in cancer research: progress and perspectives. J Hematol Oncol. 2021 Jun 9;14(1):91. doi: 10.1186/s13045-021-01105-2.
- Suvà ML, Tirosh I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol Cell. 2019 Jul 11;75(1):7-12. doi: 10.1016/j.molcel.2019.05.003.
- Chen S, Jiang W, Du Y, Yang M, Pan Y, Li H, Cui M. Single-cell analysis technologies for cancer research: from tumor-specific single cell discovery to cancer therapy. Front Genet. 2023 Oct 12;14:1276959. doi: 10.3389/fgene.2023.1276959.
- Kim N, Kim HK, Lee K, Hong Y, Cho JH, Choi JW, et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat Commun. 2020 May 8;11(1):2285. doi: 10.1038/s41467-020-16164-1.
- Sinha S, Vegesna R, Mukherjee S, Kammula AV, Dhruba SR, Wu W, et al. PERCEPTION predicts patient response and resistance to treatment using single-cell transcriptomics of their tumors. Nat Cancer. 2024 Jun;5(6):938-952. doi: 10.1038/s43018-024-00756-7.
- Chen J, Wang X, Ma A, Wang QE, Liu B, Li L, et al. Deep transfer learning of cancer drug responses by integrating bulk and single-cell RNA-seq data. Nat Commun. 2022 Oct 30;13(1):6494. doi: 10.1038/s41467-022-34277-7.
- Dohmen J, Baranovskii A, Ronen J, Uyar B, Franke V, Akalin A. Identifying tumor cells at the single-cell level using machine learning. Genome Biol. 2022 May 30;23(1):123. doi: 10.1186/s13059-022-02683-1.
- https://communities.springernature.com/posts/draft-9b0556ab-aa4d-49fd-bf3c-9c1a621defff
- Gangwal A, Ansari A, Ahmad I, Azad AK, Kumarasamy V, Subramaniyan V, et al. Generative artificial intelligence in drug discovery: basic framework, recent advances, challenges, and opportunities. Front Pharmacol. 2024 Feb 7;15:1331062. doi: 10.3389/fphar.2024.1331062.
